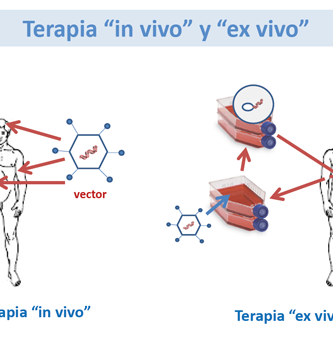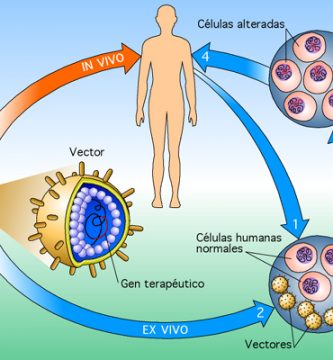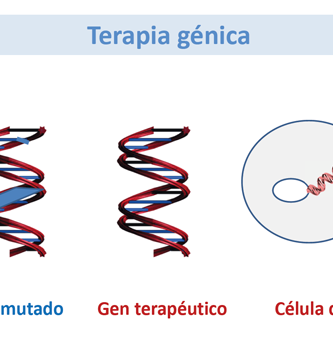
La autorización de comercialización de Glybera y Strimvelis por la Agencia Europea de Medicamentos (EMA) marcó el final del largo y a menudo problemático camino de la terapia génica desde el concepto biológico hasta la práctica médica. Glybera es un vector de virus adenoasociado recombinante (AAV) diseñado para la terapia génica de la deficiencia de lipoproteína lipasAmientras que Strimvelis es una preparación de células madre hematopoyéticas genéticamente modificadas para el tratamiento de la inmunodeficiencia combinada severaLa Administración de Drogas y Alimentos de los Estados Unidos (FDA) llenó recientemente el vacío para el mercado estadounidense al aprobar en rápida sucesión Ymlygic, un virus del herpes oncolítico modificado genéticamente, Kymriah y Yeskarta, dos formas de células T autólogas genéticamente manipuladas para la inmunoterapia del cáncer y Luxturna , un vector AAV recombinante para terapia génica de una forma rara de distrofia retiniana. Muchos más productos están a punto de seguir debido a que los ensayos clínicos fundamentales muestran eficacia y seguridad al abordar desórdenes genéticos raros así como también enfermedades neoplásicas refractarias. Sin embargo, la tecnología de la terapia génica todavía está en pañales: los procesos de fabricación están lejos de ser industrializados y los atributos de calidad y potencia mejoran continuamente gracias a la innovación y a una caja de herramientas analíticas en rápida evolución. La farmacología de la terapia génica es una nueva ciencia, aplicar conceptos desarrollados para medicamentos químicos a productos biológicos extremadamente complejos para los cuales se deben redefinir términos tales como principio activo, dosis, pureza, fuerza, toxicidad, biodistribución, eliminación, riesgo ambiental, farmacocinética y farmacodinámica de una manera creativa, aunque igualmente rigurosa . Como ejemplo, la propia definición de “fármaco” no está adaptada a las preparaciones heterogéneas de células autólogas genéticamente modificadas, que son terapias individualizadas por definición. Las agencias reguladoras de todo el mundo están tratando de hacer frente al proporcionar directrices y regulaciones destinadas a permitir el desarrollo y la comercialización de la terapia génica de una manera segura y sensata para evitar demoras innecesarias en la disponibilidad de productos innovadores para los pacientes.
Este número de Terapia molecular – Métodos y desarrollo clínicoproporciona una actualización sobre algunos problemas pendientes en la farmacología de productos de terapia génica. Seis artículos escritos por expertos en el campo abordan la fabricación de vectores y células genéticamente modificadas, interacciones vector-huésped y el uso de modelos animales para analizar la actividad, la potencia, la biodistribución, la farmacocinética, la toxicidad y la inmunogenicidad de los productos de terapia génica. En lugar de proporcionar revisiones exhaustivas, los artículos evalúan el estado del arte e identifican la controversia y las necesidades futuras en este campo en constante evolución, con referencia a las regulaciones y directrices más relevantes en Europa y los EE. UU. Este número especial tiene como objetivo crear conciencia sobre la complejidad asociada con la definición y caracterización de las propiedades biológicas y farmacológicas de los productos de terapia celular y genéticos. Vectores AAV, vectores lentivirales,
Los vectores AAV son una herramienta relativamente simple y flexible para administrar genes o ácidos nucleicos modificadores de genes a una matriz de tejidos diana para una variedad de indicaciones y son ampliamente utilizados en aplicaciones clínicas por la academia y la industria. Pennaud-Budloo y otros 4proporcionar una visión general de la tecnología actual utilizada para producir lotes de vectores AAV de grado clínico de conformidad con las buenas prácticas de fabricación (BPM) requeridas por la FDA y la EMA. Describen los dos sistemas de producción más comúnmente usados (transfección transitoria en células humanas HEK293 e infección de células de insectos por vectores de expresión de baculovirus), los desafíos asociados con la producción a gran escala y las ventajas potenciales ofrecidas por líneas celulares estables de mamíferos. Se le da mucho énfasis a la caracterización del producto, el tipo de impurezas asociadas con los diferentes sistemas de producción y purificación, y las herramientas analíticas requeridas para detectarlas de manera cuantitativa. La calidad de las partículas de AAV y la cantidad de cápsides defectuosas o “vacías” varía con los diferentes sistemas de producción y se analiza en términos de seguridad y potencia de las preparaciones de vectores. Las cápsidas vacías y su impacto sobre la respuesta inmune a los vectores de AAV son discutidas por Colella et al.5en el contexto de las consecuencias inmunológicas generales de la administración del vector de AAV a humanos. Los autores se centran en la terapia génica dirigida al hígado como un paradigma de una plataforma terapéutica que asocia la eficacia clínica probada con las interacciones específicas vector-huésped y el arma de doble filo de la inmunización / tolerización. Se presta especial atención al diseño de casetes de expresión de transgenes y nuevas cápsidas de AAV y a los estudios preclínicos necesarios para evaluar la eficacia, la potencia, la toxicidad y las respuestas inmunes humorales y citotóxicas contra vectores y productos transgénicos. El impacto de la inmunidad preexistente anti-cápside en la seguridad y eficacia de la terapia génica mediada por AAV y la correlación entre el crecimiento orgánico y la persistencia del vector a largo plazo se discuten en el contexto de los datos clínicos existentes y el futuro diseño de ensayos clínicos.6 centrándose, en particular, en los requisitos de la EMA para la autorización de ensayos clínicos en Europa. Se hace especial hincapié en la relevancia del modelo animal y los factores que determinan su idoneidad para cualquier evaluación preclínica intencionada y se describe en el contexto de los estudios de farmacología, farmacocinética y toxicología. El uso de un enfoque basado en el riesgo se describe al tomar dos ejemplos específicos como ejemplos (Glybera e Imlygic) y al analizar cómo las elecciones realizadas en los estudios preclínicos afectaron el proceso de registro de ambos productos.
Las células modificadas genéticamente son una clase diferente de productos con respecto a los vectores virales inyectables porque están hechas de un componente derivado del paciente, más comúnmente células madre hematopoyéticas o células T, y un vector viral que media la integración de un casete de expresión génica en la célula ADN. La integración, obtenida con mayor frecuencia por un vector lentiviral derivado del VIH, es un evento potencialmente genotóxico por definición, ya que interrumpe la continuidad del genoma y puede alterar (o interferir con la regulación de) los genes endógenos. La oncogénesis de inserción se ha visto en el pasado como un efecto secundario grave de las terapias génicas basadas en vectores retrovirales de la generación anterior, pero sigue siendo un problema de seguridad para cualquier vector integrante. Poletti y Mavilio 7describir la base molecular de la selección del sitio objetivo mediante retrovirus y vectores retrovirales, un conocimiento derivado de más de 15 años de investigación sobre la biología del VIH y los eventos adversos graves observados en ensayos clínicos de terapia génica. El desarrollo de la secuenciación de ADN de alto rendimiento y el análisis genómico de la estructura y configuración de la cromatina permite una comprensión detallada de la complejidad de las interacciones retrovirus-genoma, incluyendo cómo las preferencias de integración de cada virus impactan la genotoxicidad potencial de los vectores derivados de ellos. Biasco et al. 8 describen las herramientas y los análisis disponibles para analizar las características de integración y la genotoxicidad potencial de los vectores desarrollados para aplicaciones clínicas tanto en cultivo celular como in vivoen el contexto de estudios preclínicos. También describen cómo monitorear células genéticamente modificadas y su dinámica clonal en pacientes tratados. Estos análisis son todavía relativamente complejos y dependen de herramientas bioinformáticas que no están ni estandarizadas ni validadas. Sin embargo, brindan una importante lectura de bioseguridad y, en el caso de la modificación genética de las células madre de vida larga, información crucial sobre su destino y, en última instancia, sobre la eficacia de la terapia. En el caso de las células CAR-T, que están revolucionando la terapia del cáncer a pesar de su relativamente corta historia clínica, la genotoxicidad impulsada por la integración es mucho menos preocupante. Las células CAR-T tienen una vida relativamente corta y hay poca evidencia de que una célula T madura o incluso un progenitor de células T pueda “transformarse” mediante un evento impulsado por vector. Por otra parte, su eficacia depende de sus propiedades farmacológicas, que son muy diferentes de las de los vectores virales o los biológicos más convencionales. Milone y Bhoj9revise los diferentes tipos de células CAR-T actualmente bajo investigación clínica, su cinética celular después de la infusión y la relación entre las propiedades cinéticas y la actividad anticancerígena. También discuten la toxicidad asociada con las terapias de células T y la relación entre la toxicidad y la eficacia terapéutica, un aspecto intrigante aunque poco comprendido de esta forma de terapia génica.
La era de las terapias moleculares y medicinas ha llegado. Las aprobaciones iniciales recientes destacan un futuro en el que podremos tratar y curar enfermedades con tecnologías moleculares. Ahora podemos mirar hacia atrás al “punto azul pálido” de Carl Sagan que una vez fue la terapia genética y esperar las posibilidades de expansión que se avecinan. Para lograr estos objetivos, necesitaremos desarrollar una cohesión en las subespecialidades del campo (células CAR-T, ADN o virus ARN, etc.), incluidos los métodos utilizados para determinar los atributos, como principio activo, dosis, pureza, fuerza, toxicidad, biodistribución, desprendimiento, riesgo ambiental, farmacocinética y farmacodinámica, tanto en estudios con animales como con humanos. La estandarización de estos métodos proporcionará más datos que se pueden comparar directamente en todo el campo y se pueden usar para predecir resultados.