Descripción
Hay muchos enfoques diversos a la terapia génica ya que la base biológica de cada enfermedad es única, presentando un conjunto diferente de parámetros y desafíos. Sin embargo, en cada caso, se debe cumplir un conjunto básico de criterios. En primer lugar, es esencial comprender plenamente la enfermedad a tratar. Las células o tejidos asociados con la enfermedad deben estar bien definidos y accesibles. El gen y la mutación o mutaciones específicas que causan la enfermedad deben ser conocidos y debe ser posible aislar o sintetizar una copia normal y funcional de ese gen e incorporarlo en un vector. El vector entonces transfiere el nuevo gen a las células diana donde, esperanzadamente, el gen llegará a ser totalmente activo. Los papeles más comunes para el gen expresado incluyen reemplazar un gen defectuoso, inhibir o degradar un ADN, ARN o proteína deletéreos, o matar directa o indirectamente la célula.
Los trastornos génicos únicos que resultan en una pérdida de la función génica en un tejido diana específico proporcionan las opciones más fáciles para la terapia génica, aunque se han investigado estrategias para muchos tipos de mutaciones. Se ha considerado un amplio espectro de enfermedades para la terapia génica, incluyendo:
- Trastornos neurológicos, por ejemplo, enfermedad de Parkinson, enfermedad de Huntington
- Distrofias musculares
- Desórdenes inmunológicos, por ejemplo, síndrome de inmunodeficiencia combinada severa (SCIDS)
- Anomalías en la sangre, por ejemplo, talasemias, hemofilia
- Cáncer
Desafortunadamente, muchos de los trastornos más comúnmente ocurridos, incluyendo enfermedades del corazón, diabetes y presión arterial alta, son el resultado de defectos en múltiples genes que los convierten en candidatos improbables para la terapia génica usando las tecnologías existentes.
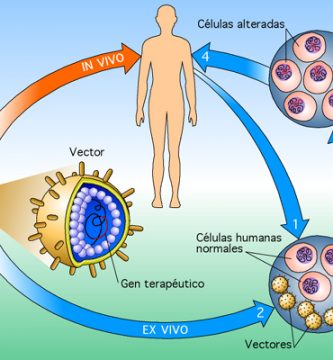
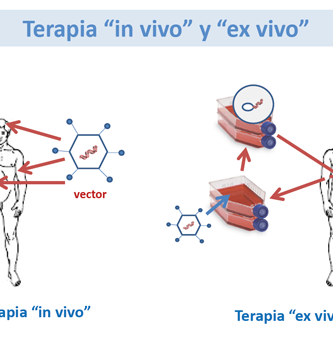
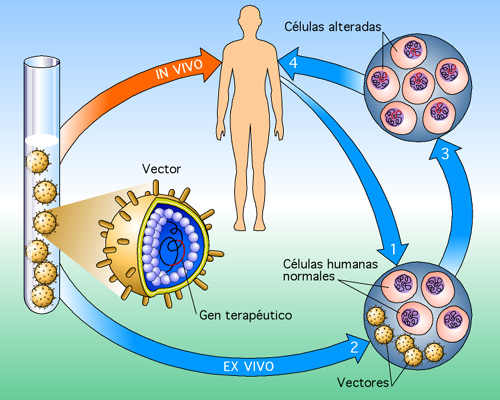
Para cada enfermedad, debe determinarse si la tecnología ex vivo o in vitro es el mejor enfoque. En la tecnología ex vivo, las muestras de células del paciente se recogen y se cultivan en el laboratorio. El nuevo gen se incorpora en las células en crecimiento, y éstas son posteriormente transferidas de nuevo al paciente. No todas las células cultivadas incluirán el nuevo gen, y no todos sobrevivirán a la transferencia. La esperanza es que un número suficiente de las células modificadas será funcional en el paciente de tal manera que la terapia revertirá la enfermedad. La terapia in vitro consiste en inyectar el nuevo gen directamente en el tejido diana donde las células individuales deben recogerlo. De los dos, este método es técnicamente más fácil y más barato, pero es más difícil determinar cuántas de las células objetivo realmente adquirir el nuevo gen. La terapia ex vivo es más costosa y requiere mucho tiempo, pero permite un mayor control de las condiciones.
Ambos procesos requieren el uso de un vector para obtener el nuevo gen a través de la membrana celular y en una célula. Los virus han demostrado ser altamente eficaces como vectores ya que éstos son entidades biológicas con una función natural de infectar células huésped. La tecnología del ADN permite que los virus sean manipulados para reemplazar la carga útil normal del material genético causante de la enfermedad con genes terapéuticos. El virus conservará su capacidad de infectar una célula huésped pero, en lugar de causar una enfermedad, depositará el nuevo gen en la célula.
También se han investigado otros mecanismos de transferencia génica. Se han desarrollado cromosomas artificiales, pero a menudo son demasiado grandes para moverse a través de las membranas celulares. Los liposomas, estructuras con membranas lipídicas, que abarcan material genético, pueden utilizarse con éxito como vectores si el liposoma es absorbido por la célula o si su membrana se fusiona con la membrana celular liberando el nuevo gen dentro de la célula.
Una vez que el gen entra en la célula, una de dos cosas ocurre. Se puede degradar y perder, lo que es un resultado desfavorable. Preferiblemente, el gen se incorporará de forma estable en el ADN de la célula diana para que pueda ser procesado como una parte normal de ese genoma. Si la terapia génica está diseñada para reemplazar un gen defectuoso, el mejor de los casos es que el nuevo gen se integre en una célula completamente renovable, como una célula madre. Teóricamente, en esta situación, el gen se incorporará permanentemente en el cuerpo del paciente y no será necesaria ninguna terapia adicional. Alternativamente, si el gen se integra en un genoma de una célula con una vida útil finita, los efectos beneficiosos del gen sólo existirán mientras que la célula vive, lo que requiere la terapia génica que se repita en un momento posterior.
Uno de los primeros éxitos de la terapia génica fue para una niña de cuatro años con deficiencia de adenina desaminasa (ADA). Esta es una forma de SCIDS que se traduce en mal funcionamiento del sistema inmunológico y puede conducir a la muerte como resultado de una infección grave. El tratamiento convencional había fallado para esta paciente, haciéndola candidata a la terapia génica. Se incorporó un gen ADA normal en un vector retroviral que transfirió el gen a los linfocitos del paciente in vitro. Las células modificadas fueron devueltas a su circulación por transfusión. Después de cinco meses, sus niveles de actividad de ADA habían aumentado de menos de 1% a 50%. Con las terapias adicionales durante los dos años siguientes, su salud mejoró mientras que la actividad de la enzima estabilizó, y ella podía comenzar una vida normal. Doce años más tarde, todavía demuestra niveles razonables de actividad de la ADA, pero la terapia génica no era una cura, ya que debe seguir recibiendo la terapia estándar de reemplazo enzimático para mantener su salud.
Enfermedades adquiridas también pueden ser tratadas con terapia génica como lo demuestra una nueva estrategia para el tratamiento del cáncer cerebral. El gen de la timidina quinasa (TK) del virus del herpes simple (HSV) tiene una propiedad enzimática que convierte el fármaco ganciclovir en una sustancia tóxica que puede matar células humanas. Se postuló que esto podría ser utilizado como una herramienta de asesinato dirigida. Para investigar, los genes clonados HSV TK se inyectaron en tumores cerebrales. En el cerebro, sólo las células tumorales se están dividiendo, por lo que estas son las únicas células que serán infectadas por el vector viral, y son, por tanto, las únicas células que recibirán el gen HSV TK. Cuando el paciente es posteriormente tratado con ganciclovir, las células tumorales que han incorporado el gen TK de HSV serán sacrificadas selectivamente. Los ensayos clínicos demostraron que las células tumorales podrían eliminarse selectivamente demostrando una reducción en el tamaño de los tumores cerebrales en siete de nueve pacientes.
Un conjunto completamente diferente de terapias es posible si la idea de terapia génica incluye el uso de ADN para el tratamiento del paciente de formas distintas a la inserción de nuevos genes en las células. Un ejemplo es el fármaco Gleevec que fue aprobado en 2001 para su uso en pacientes con leucemia mielógena crónica (LMC). Gleevec es una sustancia que se une a la proteína defectuosa producida en CML, bloqueando la actividad de esa proteína y aliviando los síntomas de la enfermedad. Esta es una terapia dirigida que afecta sólo a las células con la mutación CML, por lo que hay muy pocos efectos secundarios. La tecnología de ADN recombinante también se ha utilizado para generar copias genéticamente modificadas de vacunas (Recombivax HB), anticuerpos y productos génicos normales (insulina).
Cuidado luego del tratamiento
Si el nuevo ADN se puede incorporar de forma estable en las células diana regenerativas adecuadas, el paciente puede curarse de la enfermedad. No se requiere ningún cuidado adicional, aunque la monitorización periódica del paciente es apropiada.
Para las terapias génicas en las que el nuevo ADN se inserta en las células con una vida útil finita, el efecto terapéutico se perderá cuando esas células mueren. En estas situaciones, el paciente necesitará tratamientos continuos. La monitorización de los pacientes que reciben fármacos y sustancias derivadas de la tecnología del ADN recombinante es la misma que la terapia farmacológica estándar.
Precauciones
Actualmente, la terapia genética clásica sigue siendo experimental. Aunque muchos pacientes han demostrado una mejora significativa después de su tratamiento, al menos dos personas han muerto como resultado de este tipo de terapia. Por lo tanto, los expertos revisan cuidadosamente todos los protocolos antes de realizar cualquier estudio. La investigación inicial se realiza en un sistema modelo animal, y cualquier problema detectado se evalúa cuidadosamente antes de intentar los mismos tratamientos en seres humanos.
Riesgos
Un paciente que está recibiendo terapia génica puede enfrentar una serie de problemas potenciales. Los vectores virales utilizados pueden causar infección y / o inflamación de los tejidos, y la introducción artificial de virus en el cuerpo puede iniciar otros procesos de la enfermedad. La terapia génica funcional se basa en la incorporación estable de un nuevo gen en el ADN de un individuo. Como la integración es aleatoria, en ocasiones el nuevo gen puede insertarse dentro de otro gen que funciona normalmente, causando su daño o inactivación. Esto, a su vez, podría conducir al cáncer u otra enfermedad. Es también crítico que el nuevo gen tenga los controles reguladores apropiados de modo que el producto del gen se produzca en la cantidad apropiada. La sobreexpresión de ciertos genes puede tener resultados deletéreos. Cualquiera de estos problemas podría hacer la terapia génica ineficaz, o, en el peor, causar la muerte del sujeto.
Resultados normales
La terapia génica clásica busca tratar o curar una enfermedad definida mediante la incorporación de un gen funcional o producto génico en células diana de un individuo afectado.
Modelos de terapia génica
Las células somáticas humanas contienen 23 pares de cromosomas, y heredamos un conjunto de 23 cromosomas de cada uno de nuestros padres. Los cromosomas se construyen usando cuatro tipos diferentes de bases de ADN: adenina (A), citosina (C), guanina (G) y timina (T). El genoma humano está definido por más de 3 mil millones de pares de bases de ADN que componen nuestros 46 cromosomas; estos pares de bases de ADN están organizados en un orden específico y sirven como el modelo que distingue a los humanos de otras especies. Los estudios del genoma humano han demostrado que entre 20,000 y 25,000 genes residen a lo largo de nuestros cromosomas. Aunque la redundancia está integrada en el genoma humano, nuestro repertorio genético está exquisitamente regulado para mantener la función celular al copiar al ARN, lo que a su vez determina la formación de proteínas. Por lo tanto, incluso una sola mutación de par de bases en uno de nuestros genes puede conducir a una proteína alterada y, en consecuencia, a una enfermedad.
Aunque las complejidades de la biología humana sugieren un genoma igualmente complejo, el genoma humano es bastante similar al del ratón. El genoma del ratón está formado por 2.700 millones de pares de bases de ADN, y las estimaciones actuales sugieren que el genoma del ratón contiene al menos 28.972 genes, de los cuales, al menos, 16.927 tienen homólogos (llamados ortólogos) en humanos. De hecho, 846 enfermedades genéticas humanas se han reproducido en cepas de ratón (llamadas modelos genotípicos de ratón) que portan una mutación en el gen del ratón relacionado.
Entonces, ¿qué distingue a los humanos de otros organismos? Lo que nos hace humanos es una combinación de lo siguiente:
- La capacidad de un único producto génico (proteína) para realizar múltiples funciones debido a roles superpuestos en diferentes vías
- La capacidad de nuestros genes para someterse a diferentes tipos de empalme de ARN para producir variantes de genes con funciones alternativas
- Las funciones importantes llevadas a cabo por segmentos de ADN cromosómico localizados entre genes
Regulación epigenética
Roll Call: de los genes a la enfermedad
Debido a que heredamos un conjunto de 23 cromosomas de cada uno de nuestros padres, nuestras células somáticas contienen dos copias de cada uno de estos 20,000 a 25,000 genes. La mayor parte del genoma humano es casi idéntico de una persona a otra. De hecho, en promedio, un segmento de 1,200 pares de bases elegidas al azar de ADN cromosómico humano contiene solo un par de bases que varía entre dos individuos no relacionados. La gran mayoría de estas variantes de ADN no son perjudiciales, pero algunas alteraciones específicas en la secuencia de ADN humano están asociadas con la enfermedad.
De hecho, las estimaciones recientes sugieren que las mutaciones en al menos 383 genes humanos conducen a fenotipos conocidos, y que más de 2.336 fenotipos humanos se entienden a nivel molecular. Sin embargo, la base molecular de 1.630 fenotipos mendelianos confirmados no se conoce, y se sospecha que 2.081 fenotipos adicionales exhiben herencia mendeliana (herencia mendeliana en línea en el hombre, 2008). Aunque las “píldoras” genéticas todavía no existen para atacar la gran mayoría de las mutaciones relacionadas con la enfermedad, nuestro conocimiento de la secuencia del genoma humano ha abierto nuevas puertas para el desarrollo de enfoques terapéuticos basados en genes (Brinkman et al., 2006; O ‘ Connor y Crystal, 2006).
Tratamiento de la enfermedad antes de conocer los genes implicados
Mucho antes de que se secuenciara el genoma humano, los médicos ya estaban tratando muchas formas hereditarias de enfermedad humana con sorprendente éxito. Muchas de estas enfermedades eran trastornos metabólicos, y los médicos pudieron determinar si un producto metabólico se estaba acumulando a niveles dañinos, o si faltaba un intermediario clave en una vía metabólica, aunque aún no se conocía el gen asociado a la enfermedad. En algunos casos, dichos fenotipos de enfermedad se pueden controlar mediante la modificación de la dieta o proporcionando una proteína faltante. En otros casos, los abordajes quirúrgicos se pueden usar para reparar o reemplazar un órgano o tejido dañado por una enfermedad.
Manipulación metabólica
Los médicos han desarrollado enfoques para regular las vías metabólicas asociadas con una serie de trastornos, como la fenilcetonuria (PKU), la anemia de células falciformes, el angioedema hereditario, la hipercolesterolemia familiar, la talasemia y muchos otros. A menudo, esta forma de “manipulación metabólica” puede lograrse modificando la dieta de un paciente. Por ejemplo, los pacientes con PKU acumulan altos niveles de un bloque de construcción de proteínas, llamado fenilalanina, en su torrente sanguíneo. Los médicos pueden diagnosticar PKU usando un simple análisis de sangre con punción en el talón para detectar niveles altos de fenilalanina a los tres días de edad. Una vez diagnosticados, los bebés con PKU reciben una dieta baja en proteínas y fenilalanina, lo que ayuda a prevenir el deterioro cognitivo asociado a la PKU.
En otros casos, la manipulación metabólica implica el uso de pequeñas moléculas o fármacos para dirigir la actividad de las proteínas relacionadas con la enfermedad. Por ejemplo, la hipercolesterolemia familiar se asocia con altos niveles de colesterol “malo” (LDL) y enfermedad cardíaca temprana; en este caso, el tratamiento incluye tanto modificaciones dietéticas (una dieta baja en colesterol) como la administración de una clase de medicamentos llamados estatinas.